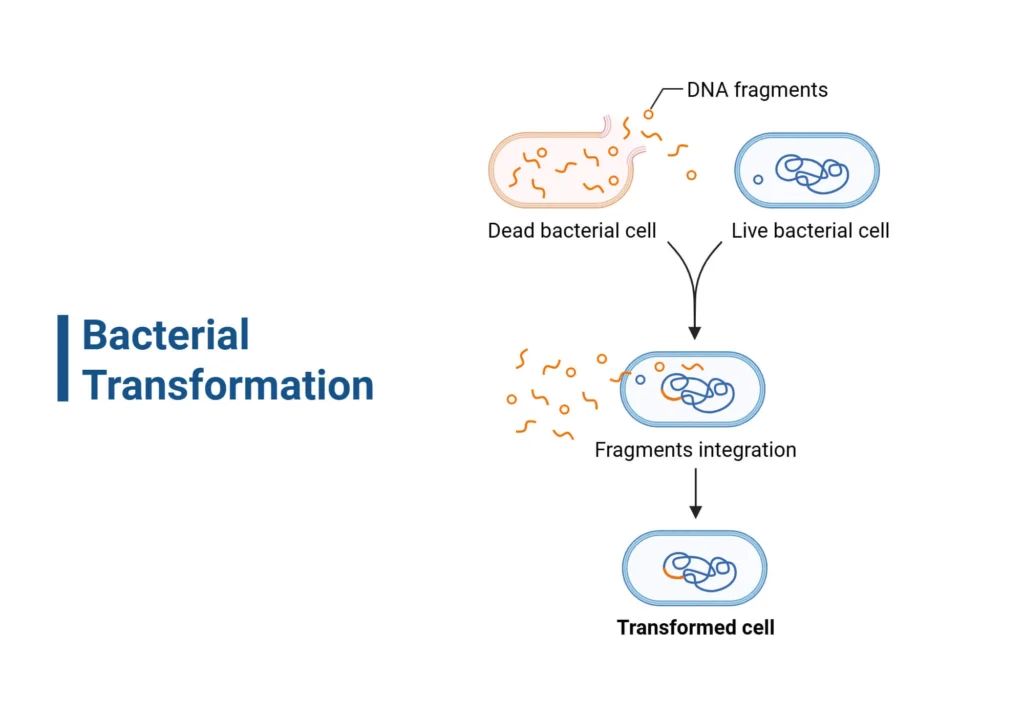
Bacterial transformation is a process by which a bacterial cell takes up exogenous foreign DNA from its environment, leading to a stable hereditary change.
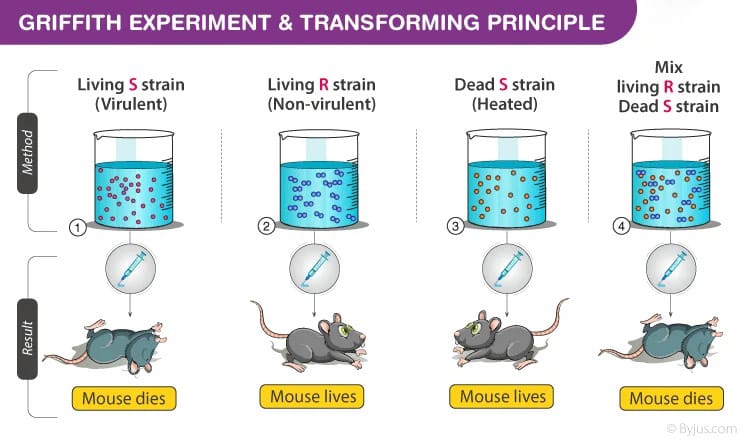
The phenomenon of transformation was first demonstrated by Frederick Griffith in 1928, during his experiments with Streptococcus pneumoniae, where he observed the transfer of virulence genes between bacterial strains.
Types of Bacterial Transformation
Transformation in bacteria can occur in two different ways: naturally or artificially
Natural transformation
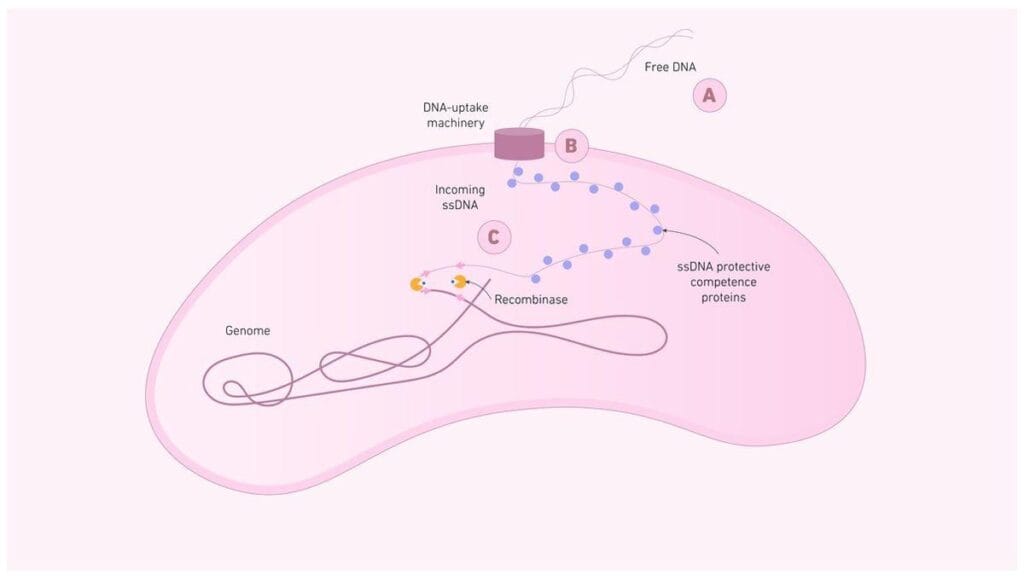
Genetically determined process by which certain bacteria can naturally take up and integrate exogenous DNA from their environment. Eg, Streptococcus, Bacillus, Haemophilus, Neisseria
Artificial transformation
It is the process by which bacterial cells are artificially made competent (either physically or chemically) in the laboratory, enabling them to take up exogenous DNA from the environment. Eg, Escherichia coli requires artificial induction in the laboratory to become competent. The first demonstration of artificial, calcium-dependent DNA transfer into E. coli was provided by Mandel and Higa (1970).
Bacteria mainly take up plasmid DNA (circular and self-replicating) in laboratory transformation; linear DNA is usually degraded and rarely maintained.
Bacterial transformation is a fundamental technique in molecular biology, which has various applications including gene cloning, routine DNA manipulation and maintenance, in vivo diagnostics, gene therapy, antibiotic resistance studies, metabolic engineering, cDNA Library Construction, Routine DNA Manipulation and Maintenance, Advanced Selection and Screening Techniques (such as Blue-white screening), Testing Novel Delivery Systems, Microfluidic Systems, etc.
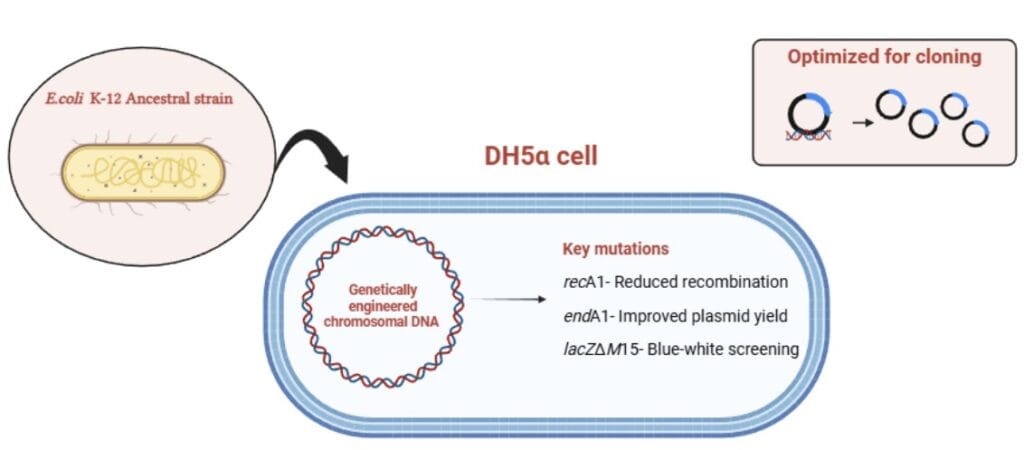
Why is DH5α used for transformation?
DH5α is a genetically engineered Escherichia coli K-12 strain, which is a widely used laboratory strain in molecular biology to amplify exogenous plasmid DNA. DH5α is the Gold Standard for cloning because of its high transformation efficiency. Escherichia coli DH5α strain is genetically optimized for recombinant DNA work.
Highly competent commercial DH5α cells typically achieve efficiencies of 109 colony-forming units (CFU) per microgram (μg) of plasmid DNA.
Key genetic features responsible for high transformation efficiency include:
recA1 – This mutation inactivates the recA protein, which results in the reduction of homologous recombination, ensuring the stability of foreign plasmids without rearrangement or degradation.
endA1– This mutation inactivates a non-specific endonuclease that leads to significantly higher quality DNA minipreps. In the wild type, the enzyme often degrades plasmid DNA during the isolation process.
hsdR17(rK−,mK+) – This is the mutation of the restriction-modification system of E. coli. With this mutation, the cells do not digest the foreign DNA (restriction negative/ rK−) while they can still be able to methylate its own DNA, protecting it from accidental digestion (modification positive/ mK+).
ϕ80dlacZΔM15 – Phage DNA with modified lacZ, which involves a specific modification where the lacZ gene is 94 bases shorter at the N-terminus than in the parental strain, enabling α-complementation for blue-white screening to easily identify bacteria carrying a plasmid.
The Principle Behind Transformation
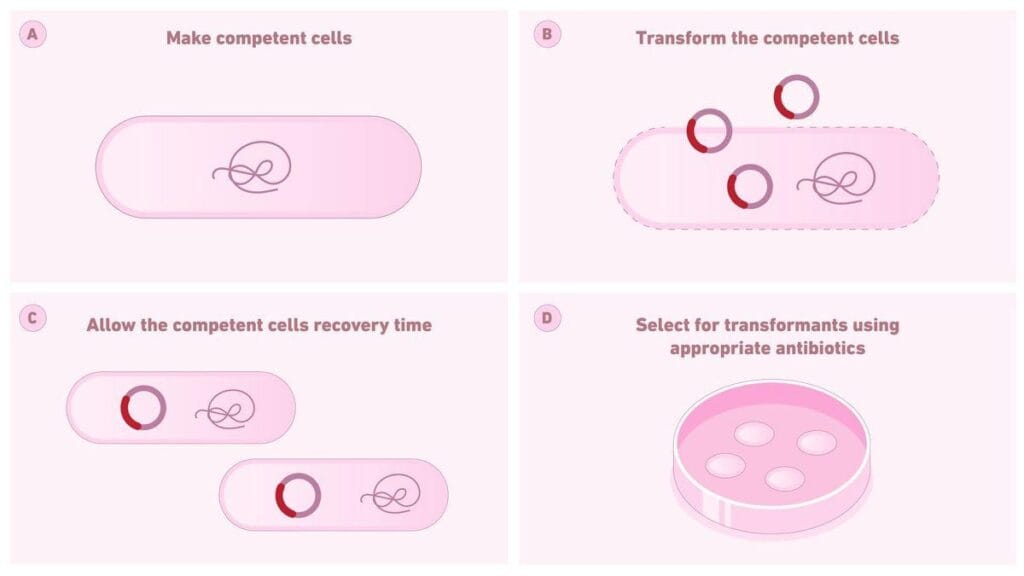
The principle behind transformation involves overcoming the biological barriers of bacterial cells through physicochemical manipulations, making the cell competent, which enables it to take up foreign extracellular DNA. In strains like Escherichia coli DH5α, transformation is generally not natural and must be induced through an artificial method.
The basic principle of transformation can be studied in three main steps:
Induction of competence: Competence is a state where a bacterial cell gains the ability to take up foreign DNA. The basic principle of competence is charge neutralization and membrane alteration. Both DNA and the bacterial cell membrane are negatively charged, which results in electrostatic repulsion. In the case of the chemical method, divalent cations (Ca2+) bind to the negatively charged phosphate groups as well as other groups ( such as hydroxyl and carboxyl) on the cell membrane, causing a neutralization effect. This neutralization results in the formation of a chemical bridge that promotes contact between exogenous DNA and the cell membrane.
DNA uptake: Once DNA is adsorbed on the cell membrane, it must cross the cellular envelope into the cytoplasm. This is achieved through thermal imbalance or heat shock. Cells are typically kept at 0°C, shifted rapidly to 42°C, and then returned to 0°C, which induces the transient opening of gated membrane channels, allowing DNA to enter the cytoplasm. Unlike chemical methods, electroporation does not involve DNA binding to the cell surface. Instead, the intense electric field causes transient permeabilization of the cellular membrane, forming pores. As a result, the exogenous DNA is driven through pores by electrophoretic force.
Recovery and stabilization: Once the foreign DNA is inside the cell, it needs to be established as a stable genetic material. Transformed cells are often fragile due to membrane damage. A post-incubation period at 37°C in a nutrient-rich medium (like SOC) allows the cells to repair their membranes and express antibiotic resistance markers before being plated on selective media.
Essential Reagents for Bacterial Transformation
The essential reagents required for bacterial transformation depend on the method employed during competent cell preparation ( either electroporation or chemical method).
Category
Reagent/ Medium
Method
Role in transformation
Genetic material
Plasmid DNA
Chemical and electroporation
A vehicle for carrying a foreign gene into the host cell.
Recipient cell
Competent E. coli (such as DH5α, DH10B, JM109, XL1-Blue)
Chemical and electroporation
Host cells are prepared to uptake foreign DNA
Growth media
LB (Luria–Bertani)
Chemical and electroporation
A standard broth used for initial starter cultures.
Recovery media
(SOC) Super Optimal broth with Catabolite repression- Contains 2% Bacto tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, and 10 mM MgSO4 supplemented with 20mM glucose.
Chemical and electroporation
Used during the one-hour recovery (outgrowth) phase to help cells recover and express antibiotic resistance.
The transformation of DH5α Cells can be done by various methods (electroporation and chemical methods being the most commonly used in laboratories).
Protocol for the chemical method of transformation:
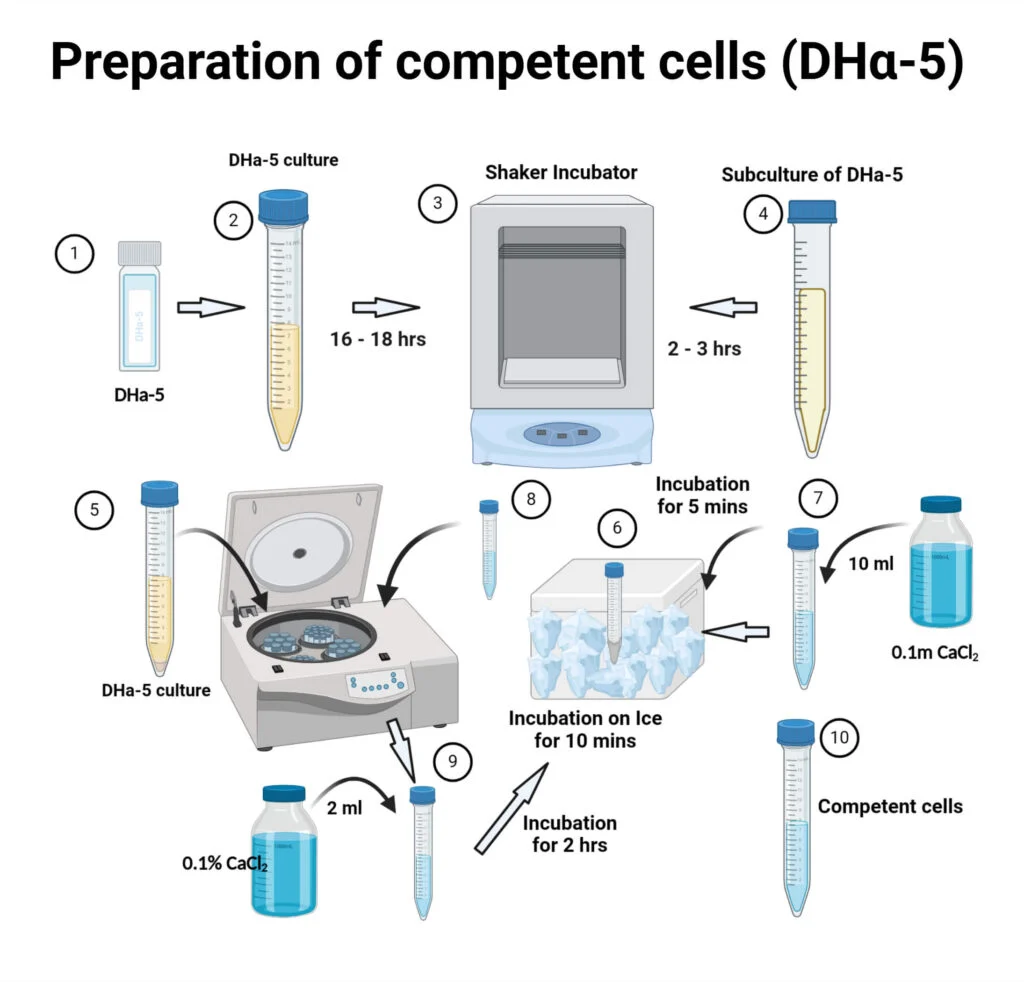
Streak DH5α cells from a frozen glycerol stock onto a fresh LB or SOB agar plate and incubate overnight at 37°C (16-18 hours).
Take a single well-isolated colony from the plate and inoculate it into 2-5 mL of SOB/LB medium and incubate at 37°C overnight with vigorous shaking at 200 rpm.
Inoculate 50 mL of freshly prepared LB /SOB broth with 500 µL of the overnight starter culture and incubate at 37°C (200 rpm) until the culture reaches an optical density (OD of 0.5. This typically takes about 4 hours.
Immediately place the culture on ice for 10 minutes to stop growth.
Spin the cells at 2,000 x g for 10 minutes at 4°C.
Discard the supernatant. Gently resuspend the pellet in 15 mL of ice-cold 0.1 M MgCl2
Incubate the mixture on ice for 30 minutes.
Centrifuge the cells at 4,000 rpm for 8–10 minutes at 4°C.
Discard the supernatant. Gently resuspend the final pellet in 4 mL of 0.1 M CaCl2 containing 20% (v/v) glycerol and aliquot 100 μL in different epitubes, store at -80°C.
For transformation, thaw the competent cell from -80°C on ice.
Mix the cells with 1 μL of plasmid and incubate on ice for 30 minutes.
Subject the tubes to a 42°C water bath for 45 seconds.
Immediately place the tubes back on ice for 2 minutes
Add 1000 μL of SOC media and incubate at 37°C for 1 hour with shaking
Plate the entire volume onto LB agar containing the appropriate antibiotic and incubate at 37°C overnight.
Protocol for the electroporation method of transformation:
Streak DH5α cells from a frozen glycerol stock onto a fresh LB or SOB agar plate and incubate overnight at 37°C (16-18 hours).
Take a single well-isolated colony from the plate and inoculate it into 5 mL of SOB/LB medium and incubate at 37°C overnight with vigorous shaking at 200 rpm.
From the O/N culture, inoculate 900 ul in 50 mL of fresh LB media ( secondary culture) and incubate at 37 °C with shaking at 200 rpm.
Check OD of the secondary culture until it becomes 0.4 to 0.5.
Once the appropriate OD is reached, keep the grown culture on ice for 15 mins.
Centrifuge the cells at 4000 rpm at 4°C for 10 mins.
Discard the supernatant and resuspend the pellet in 15 mL sterile cold distilled water.
Centrifuge at 4000 rpm for 10 mins at 4°C.
Repeat the washing step with sterile water.
Discard the supernatant and resuspend the pellet in 1 ml of ice cold 10% glycerol.
Centrifuge at 4000 rpm for 10 mins at 4°C.
Discard the supernatant and resuspend in 900 ul of 10 % glycerol.
Aliquot 50ul in different epitubes and store at -80°C.
For transformation, add 2 ul plasmid in one of the aliquoted 50 ul competent cells epitube.
Mixture transferred to pre-chilled cuvette.
Electric shock at 1.79 kV in an electroporator.
Add 1 ml of SOC media immediately.
Transfer to sterile epitubes.
Incubate at 37 °C for 1 hour.
Spread plate on LB containing appropriate antibiotic media and incubated at 37 °C overnight.
Optimizing the Heat Shock
Optimization of the heat shock step is essential for maximizing transformation efficiency in DH5α.
To achieve the highest transformation efficiency in DH5α, the optimal temperature for heat shock is 42°C, and the optimal time is between 30 and 45 seconds.
Following the heat shock, tubes must be transferred immediately to an ice bath. Protocols typically recommend a 2-minute incubation on ice to stabilize the membranes.
After the incubation for 2 mins, the addition of SOC or SOB medium (which contains glucose and magnesium salts) is vital, and the cells should be incubated at 37°C with vigorous shaking for approximately one hour to allow for the expression of antibiotic resistance before plating.
The heat shock should be performed without agitation.
The material of the transformation tube can impact the heat transfer during the heat shock step. Using polypropylene tubes is recommended over glass tubes. Glass tubes can lead to a tenfold decrease in competence, likely due to different thermal conductivity properties affecting the heat pulse.
Standardizing competent cell volume (commonly 100 µL) during heat-shock optimization ensures consistent heat transfer and reliable comparison of transformation efficiency.
The Recovery Phase
The recovery phase (outgrowth period) is an essential step of transformation after heat shock or electric pulse, where the transformed bacteria are incubated at 37°C in nutrient-rich media with vigorous shaking.
Purpose of the recovery phase:
Antibiotic resistance expression: Bacteria need time to transcribe and translate the resistance genes (such as ampicillin or kanamycin resistance) provided by the plasmid. Without this period, the cells would be killed upon contact with selective agar plates.
Cellular repair: For methods like electroporation or heat shock, the recovery phase allows cells to recover from the physical stress.
Maximizing Efficiency: It is vital to shake the tubes vigorously (typically between 200 and 220 rpm) during this hour to ensure adequate aeration and nutrient access for the recovering cells.
The entire suspension, or a desired portion, is spread onto agar plates containing a specific antibiotic, such as ampicillin (at concentrations of 50–100 µg/ml), and the plates are then incubated overnight at 37°C to allow transformed colonies to develop.
Plating and Selection
Plating and selection is the final stage of transformation, where the bacterial cell population is transferred to a solid agar medium containing a selective marker (such as ampicillin). Only the transformants that have taken up the plasmid can grow on the plate and form colonies.
The selection is typically performed on LB agar plates, and the choice of antibiotic depends on the resistance marker of the plasmid.
Plates are incubated overnight at 37°C, and after incubation, bacterial colonies are counted manually to calculate the transformation efficiency (TrE).To verify the process, controls are often performed by transforming cells with water instead of DNA; these should yield zero colonies.
Table1: Plating & Selection Parameters for transformed DH5α
Parameter
Standard Protocol Requirement
Media Type
1.5% LB Agar
Antibiotic (Example)
Ampicillin (50–100 µg/ml)
Incubation Temperature
37°C
Incubation Time
Overnight
Plating Volume
100 µl to the entire 1 ml suspension
Calculating Transformation Efficiency
Transformation Efficiency (TrE) is a quantitative measurement used to determine how many bacterial cells successfully took up a plasmid during a transformation protocol. It is expressed as colony-forming units per microgram (cfu/µg) of plasmid DNA. By definition, transformation efficiency is the number of colony-forming units produced by transforming 1 µg of plasmid DNA into a given volume of competent cells.
TrE =Number of colonies (cfu)Amount of DNA used (µg) ×Dilution Factor
Where,
Number of Colonies (cfu): The manual count of bacterial colonies growing on the selective agar plate
Amount of DNA used (µg): The mass of the plasmid added to the competent cells, converted to micrograms
Dilution Factor: This accounts for the fraction of the total recovery mixture that was actually spread on the plate
Example: Calculation of Transformation Efficiency (TrE)
Given:
Amount of plasmid DNA used = 10 ng
Total recovery volume after transformation = 1000µL
Volume plated on LB + antibiotic plate = 100 µL
Number of colonies observed on the plate = 120 colonies
Step 1: Convert the DNA amount to micrograms
10 ng=0.01 µg
Step 2: Calculation of dilution factor
Since 100 µL was plated,
Dilution factor = 1000/100 = 10
Step 3: Apply the transformation efficiency formula
TrE =Number of colonies (cfu)Amount of DNA used (µg) ×Dilution Factor
=1200.01 × 10
= 120,000 cfu/µg
Hence, transformation efficiency = 120,000 cfu/µg
Controls in Transformation: Positive vs. Negative
Control type
Component added
Purpose
Expected outcome
Negative
Competent cells + waterUntreated cells + DNA Competent cells + DNA (no pulse/shock)
Verify antibiotic efficacy or lack of contamination, ensure competency induction is required for uptake, confirms the physical trigger is necessary for transformation
Zero colonies
Positive
Competent cells + standard plasmid (such as pUC18 or pBR322)
Calculate and verify transformation efficiency
High colony count (e.g., 107–109 cfu/µg
Troubleshooting Common Failures
In E. coli, supercoiled and circular DNA transform much more efficiently than linear DNA. TrE is stable for plasmids up to 7 kb but drops to 15%–46% for plasmids between 10 kb and 17 kb.
Transformation efficiency is highly dependent on the quality of the chemicals used. Buffers should be prepared fresh on the day of use, and the pH must be precisely adjusted (e.g., pH 5.8 for Buffer I).
Competent cells with low transformation efficiencies can cause few or no growth on the plates. To solve this, the transformation efficiency of an uncut plasmid of known concentration, such as pUC19, can be calculated. If the transformation efficiency is lower than <104 cfu/μg DNA, the competent cells need to be remade, or commercially available high-efficiency competent cells are recommended.
The most common problem during electroporation is the electrical sparking due to high salt contamination from the DNA or the growth media. In order to solve this, the cells must be washed extensively with 10% glycerol or water.
High concentrations of plasmid DNA can have a toxic effect on cells, decreasing overall efficiency. The optimal amount for DH5α is often around 5–10 ng.
The plasmid size can also affect the transformation efficiency. Transformation using a small plasmid has higher efficiency as compared to a larger plasmid. To transform a larger plasmid, the right competent cells need to be used, and the electroporation method can also be employed for higher efficiencies.
Temporary pores are formed in the cell membrane of transformed cells due to the heat-shock step or electric pulse, which ultimately leads to stress. To recover the cells from this stress, a nutrient-rich medium, such as SOC medium (Super Optimal Broth with Catabolite Repression Medium), is required. SOC medium is preferred over LB because it has richer nutrients, magnesium ions, and glucose that promote faster cell repair, plasmid establishment, and antibiotic resistance gene expression, resulting in higher transformation efficiency.
Temperature also plays a key role during the bacterial transformation, especially while working with chemically competent cells. Any deviation from the standard heat-shock protocol can negatively affect the transformation efficiency. Following transformation, the cells need to be incubated at an optimal growth temperature (37 °C), in order to recover the cells and the antibiotic resistance gene. This recovery can be performed in a shaking incubator so that the nutrients get evenly distributed in the growth medium.
Incorrect antibiotic or antibiotic concentration can affect the number of colonies on the plate after transformation. Using the wrong antibiotic causes no colonies on the plate, and too low a concentration of antibiotic causes satellite colonies (non-transformed bacterial colonies on the plate forming a uniform layer). The selectable marker in the plasmid needs to be checked properly before performing the transformation. An appropriate concentration of antibiotic needs to be used while performing the transformation.
Conclusion
Bacterial transformation (mainly Escherichia coli DH5α cells) is a fundamental technique in molecular biology, particularly for cloning and plasmid propagation. DH5α serves as an ideal host for maintaining recombinant DNA because of the modifications, such as reduced recombination activity and enhanced plasmid stability. A clear understanding of the transformation principle, along with careful optimization of critical steps like competence induction, heat shock, recovery, and selection, is essential for achieving high transformation efficiency. Proper use of controls and troubleshooting strategies further ensures the reliability and reproducibility of results. Overall, DH5α transformation not only facilitates successful cloning experiments but also provides a strong technical foundation for advanced applications in genetic engineering, functional genomics, and biotechnology research.