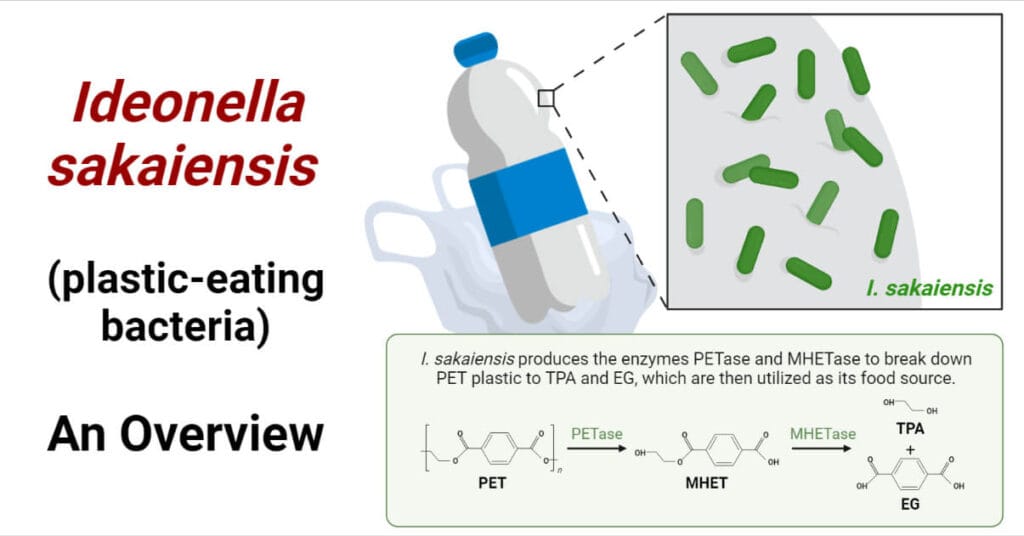
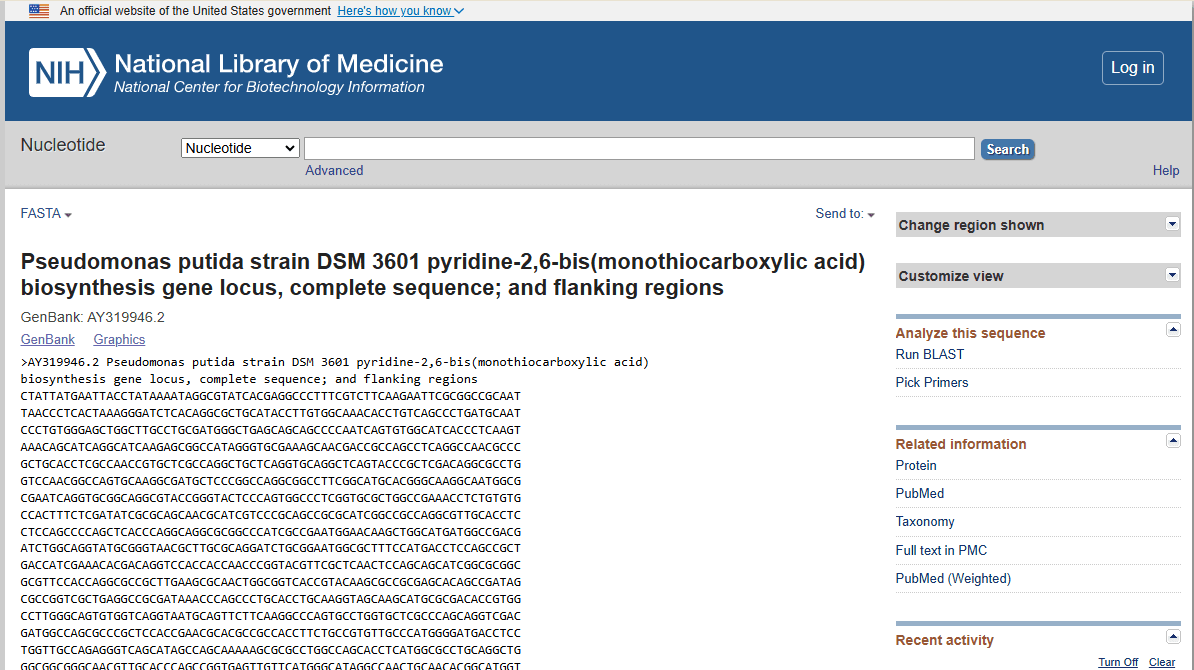
FASTA (Fast-All) is a pioneering sequence alignment tool developed by David J. Lipman and William R. Pearson in 1985. It enables the comparison of nucleotide or protein sequences against existing databases to identify similarities, facilitating insights into sequence function and evolutionary relationships.
Key Features of FASTA:
Sequence Alignment: Compares query sequences with database sequences to find regions of similarity.
Versatility: Applicable to both nucleotide and protein sequences.
Heuristic Approach: Employs a rapid, heuristic algorithm to identify potential matches efficiently.
FASTA Programs:
FASTA has evolved into a suite of programs tailored for specific sequence comparison tasks:
FASTA: Compares DNA to DNA or protein to protein sequences.
SSEARCH: Utilizes the Smith-Waterman algorithm for precise protein-protein or DNA-DNA comparisons.
FASTX/FASTY: Aligns DNA sequences to protein databases by translating DNA into protein sequences, accommodating frameshifts.
TFASTX/TFASTY: Aligns protein sequences to DNA databases, translating DNA sequences in all six reading frames.
Working Mechanism of FASTA:
Identifying Regions:
The query sequence is divided into smaller words called k-tuples (ktup).
A lookup table is created to identify matches between k-tuples in the query and database sequences.
Regions with high similarity are represented as diagonals in a matrix, with the top ten regions selected for further analysis.
Re-Scoring:
The top ten diagonals are rescored using scoring matrices (e.g., BLOSUM50 for proteins).
High-scoring subregions within these diagonals, known as initial regions, are identified.
Joining Threshold:
A score cutoff is applied to exclude segments unlikely to be part of the final alignment.
Remaining initial regions are joined to form a longer alignment.
Final Alignment:
A dynamic programming algorithm refines the alignment, producing the final result.
Applications of FASTA:
Sequence Annotation: Identifying functional regions by comparing sequences to known databases.
Evolutionary Studies: Assessing homology and evolutionary relationships between sequences.
Gene Identification: Detecting genes within genomic sequences through similarity searches.
Protein Function Prediction: Inferring protein function based on sequence similarity to characterized proteins.
FASTA Format:
The FASTA format is a text-based representation of nucleotide or protein sequences, beginning with a single-line description starting with a ‘>’ symbol, followed by lines of sequence data.
Advantages of FASTA:
Speed: Heuristic approach allows for rapid sequence comparisons.
Simplicity: Straightforward format and operation make it accessible for various applications.
Versatility: Applicable to a wide range of sequence types and comparison needs.
Limitations:
Approximation: Heuristic methods may miss some alignments compared to exhaustive algorithms.
Sensitivity: Less sensitive than methods like the Smith-Waterman algorithm for detecting weak similarities.
Conclusion:
FASTA remains a fundamental tool in bioinformatics for sequence alignment and database searching. Its development marked a significant advancement in computational biology, providing researchers with a means to rapidly compare biological sequences and infer functional and evolutionary insights. Despite the emergence of newer tools, FASTA’s speed, simplicity, and versatility continue to make it a valuable resource in the field.









: Revolutionizing Pharmaceutical Discovery")